
What longevity genetics is missing
Allosomal aneuploidy biobanks; a modest proposal to fill in data gaps

Note that this is posted in a personal capacity and this blog post does not represent any positions of Relation. Special thanks to: 1.) Madi Ueland who pointed me in the direction of loads of literature I wasn’t aware of, and 2.) Norn Group who invited me to speak on what’s happening in the world of AI for drug discovery & longevity.
Human longevity is substantially heritable — recent estimates put intrinsic heritability near 50% once extrinsic deaths (accidents, infection, violence) are accounted for — yet genome-wide association studies (GWAS) explain very little of it, aka “missing biology”. One of the largest under-exploited clues sits in plain sight: sex, but despite various sociological explanations (violence, risk-taking behaviour) and broad-brush biological explanations (X-linked genetic disorders, immune function, infection risk), we do not yet have a molecular (consensus) understanding for why women live longer than men. This effect is observable at all stages of life, even in utero.
From a gene regulatory perspective, we expect the variation to be driven by development, immune function, endocrine signalling, and genetics — confounded by chromosomal structural differences. However, human longevity GWAS have only produced a small number of sensible signals relative to the size and importance of the phenotype. Several of the few replicated signals even act sex-specifically (APOE’s effect is stronger in women, and equivalently for FOXO3 in men), so pooling the sexes can blur real effects. I would argue that one of the main reasons for this “missing biology” is that we treat sex as a single covariate to be adjusted away, rather than modelling the two biological axes it bundles: developmental effects and sex-chromosome gene dosage effects. And, given that drugs often mimic the effects of genetic variation (with different effect sizes, acting across different timescales), if we truly want longevity drugs, then understanding the genetic basis for differences in life expectancy is a natural starting point.
For an analogy, consider how we use the body mass index (BMI). BMI is defined as a person’s weight divided by the square of their height (kg/m²), which gives a single number that combines two distinct measurements. Because it forces a fixed relationship between height and weight, BMI can obscure associations that height or weight would each reveal independently. In short, two people with very different builds can share an identical BMI. Sex shares some similarities to the BMI example above.
Allosomal aneuploid populations as counterfactuals
Sex is an awkward categorical variable to incorporate into statistical analysis. Neither “male” nor “female” are single molecular exposures, each bundle together (at least) two major biological axes. First, sex determination in mammals is linked to the Y-chromosome gene, SRY, that initiates testis determination and downstream male sexual development, creating lifelong differences in anatomy, hormone profiles, immune function, metabolism, etc. The second is sex-chromosome dosage: XX and XY cells differ even after accounting for developmental differences. Although one X chromosome is largely inactivated in XX cells, a meaningful fraction of X-linked genes escape X inactivation. Reviews estimate that roughly ~15–30% of human X-linked genes may escape inactivation. These escape genes exhibit dosage-sensitive biology that could plausibly contribute to sex differences in ageing.
When we collapse these axes of variation, we then become unable to ask very fundamental counterfactual questions: what is the phenotype of a man with more expression from X-linked escape genes? What is the phenotype of a woman with one fewer X chromosome?1
However, human counterfactuals already exist in the form of Klinefelter and Turner syndrome. Klinefelter syndrome (KS), or “47,XXY”, are males with an extra X chromosome. Turner syndrome (TS), or “45,X”, are females with partial or complete loss of a second sex chromosome. Other rarer sex aneuploidies like 47,XXX and 47,XYY also exist, see below for life expectancy estimates (as known). Whilst these patients often suffer from chronic disease (e.g., type 2 diabetes and metabolic syndrome, cardiovascular disease, osteoporosis, and autoimmune thyroid disease) and infertility, they are mostly healthy but with a reduced life expectancy. They are also not vanishingly rare. Klinefelter syndrome affects about 1 in 600-660 male births and is substantially underdiagnosed; many people are only identified during infertility assessments. Turner syndrome is reported in roughly 1 in 2,000-2,500 live female births, with wide phenotypic variability, especially in mosaic forms. Compared to other “age-associated” rare diseases (progerias, Werner syndrome, Cockayne syndrome etc) which implicate only a handful of genes (mostly relating to DNA repair and genome integrity), these aneuploidies of the sex chromosomes have much broader applicability.2 Their effects are not uniformly harmful either: Klinefelter syndrome is associated with reduced prostate-cancer risk and Turner syndrome with reduced breast-cancer risk, pointing to protective dosage effects for specific age-related diseases.

Complex emergent trans-regulation
Molecular characterisation of Klinefelter and Turner syndromes remains sparse relative to common-disease biobanks. Reanalysing a 2020 study that measured PBMC (white blood cell) bulk RNA expression of a KS and TS patients with age-matched 46,XX and 46,XY euploid controls, we see a clear dose-response relationship for genes on the X and Y chromosomes in relation to copy number. In effect, these aneuploidies act as natural dosage dials; by tuning copy number up and down rather than switching genes off, much as a drug modulates rather than abolishes its target.3
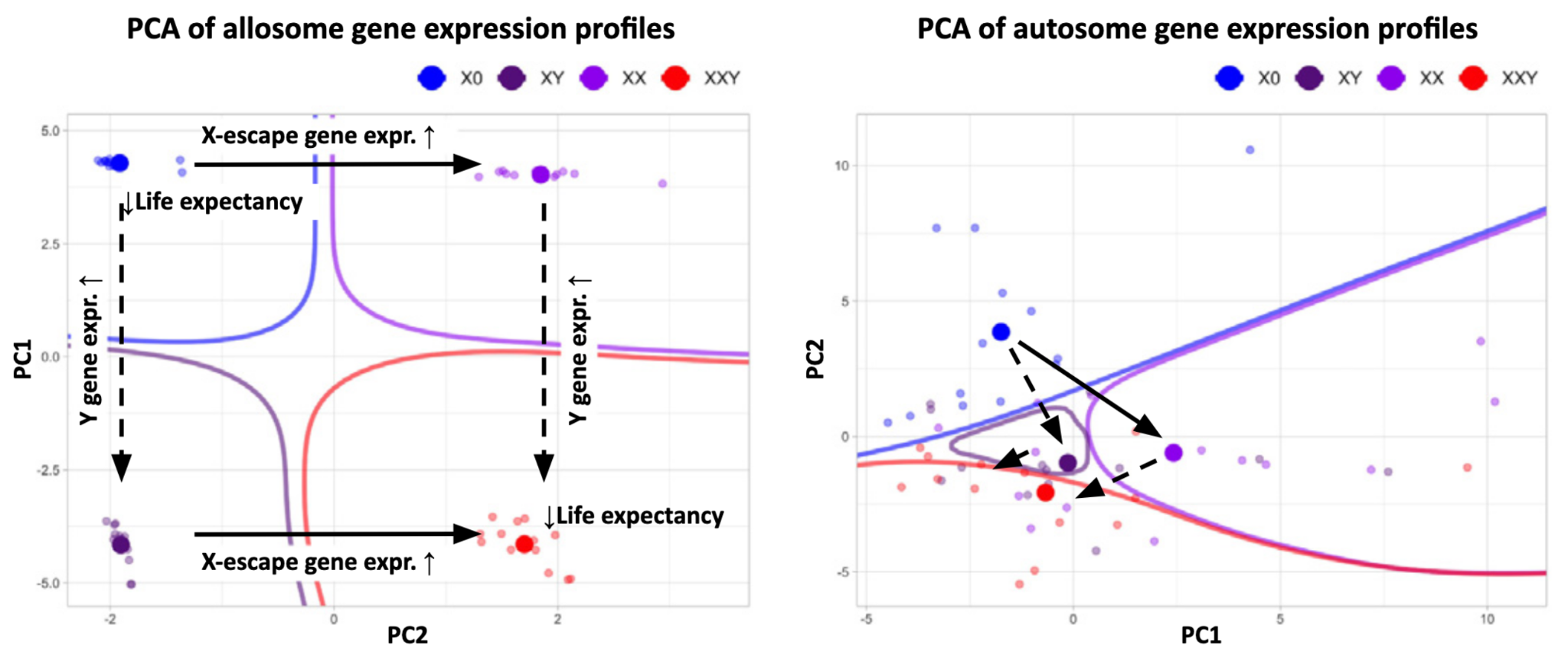
However, much more complex relationships emerge when examining the autosomes, hinting at significant perturbations to human physiology. When examining known genes implicated in longevity, we observe dysregulated gene expression. For example, similar to some autoimmune conditions, Turner syndrome women have higher LPL expression than 46,XX women (suggesting one possible plausible reason for lower life expectancy). Several newer molecular studies now support this broader picture of autosomal gene dysregulation, including transcriptomic, epigenomic, single-cell, metabolomic studies. However, these datasets remain small, tissue- or development-stage-specific, and largely cross-sectional or disease-mechanism-focused, rather than longitudinal, population-scale resources designed to connect sex-chromosome dosage to ageing and survival.
How biobanks help establish causality and reversibility
To discover drug targets likely to treat complex disease, we typically look for characteristics that suggest the drug target is not entirely dissimilar from the targets already manipulated by successful drugs, e.g., expressed in the relevant cell type, druggable from a structural perspective, etc. Most notably, the retrospective study by Nelson et al, showed that when you consider drug targets associated with clinical success, they are ~2.6 times more likely to have some genetic basis for a disease-associated phenotype related to them, often via GWAS. And because germline genetics are broadly assumed to be immutable, then they are causally upstream and are driving disease risk.4 However, causality is not the same thing as reversibility, so we need to build model systems (either in vitro or in vivo) that mimic key parts of pathophysiology, and show that a healthy phenotype can be restored. Biobanks help with both problems because whilst we typically study germline genetics from a blood draw, we may also retain tissue samples that can be used to either study cellular architecture (static) or build dynamic cellular models of disease.
The biobank evidence we already have shows the gap. In the UK Biobank, for KS researchers identified 213 men with 47,XXY and 143 with 47,XYY among 207,067 men of European ancestry, and of the 244,848 women over 40, they found 30 women with 45,X, 186 with mosaic 45,X/46,XX, and 110 with 47,XXX. Because biobanks detect aneuploidies by screening everyone’s genome rather than relying on clinical referral, they capture undiagnosed and mild cases that clinic-based cohorts miss.5 However, these numbers are still shockingly small to be meaningful from the perspective of a population genetics statistical analysis.
So, what do we need to do? We need to build comprehensive data resources that link genetic variation, karyotype status, single-cell or spatial omic profiling of important tissues (e.g., white blood cells to characterise the immune system etc) across thousands of individuals with KS and TS. This can be done by extending the UK Biobank or through entirely new initiatives. Taking a step further than what the UK Biobank already does, we should also collect skin fibroblasts for development of iPSC cell banks to build models of relevant disease processes, and track individuals over time to understand organs and tissues as they accumulate damage.
In closing
Much of modern longevity genetics still leans on finding putative ageing genes across species (not within humans). Human studies are possible (consider centenarian, familial studies beyond standard GWAS) but have yielded few robust loci relative to lifespan’s heritability. We know from drug discovery writ large how challenging preclinical translation can be (i.e., are you actually testing the same biology across experimental systems?). Whilst there are highly conserved biological processes across mammals (e.g., bone formation, cartilage destruction, muscle contraction etc), many of the processes that we are most interested in have diverged over evolutionary timescales, e.g., immune function. For example, humans, wild mice, and standard laboratory mice have very different pathogen ecologies (sterility of environments, exposure to airborne pathogens vs. bacteria/fungal colonies), and laboratory husbandry can strongly shape phenotype. Moreover, the lab animals we experiment on often are bred in artificial conditions skewing their phenotypes in unexpected ways: telomere length varies substantially across mouse species and strains; some established laboratory strains have unusually long telomeres, while wild-derived strains can be shorter or longer depending on background.
Human longevity genetics has spent years looking for variants that explain why some people live exceptionally long lives. That remains important. But perhaps one of the largest natural experiments in human survival has been sitting in front of us the whole time: women live longer, girls survive better even in infancy, and sex chromosomes should not be just binary labels in a covariate table. If human genetics is one of the best maps we have for drug discovery, then allosomal aneuploidy biobanks could help draw a missing section of the longevity map.
This blog post has been adapted from a perspective in Trends in Genetics.
In mice, the “four core genotypes” model already decouples aspects of this in mice (gonadal sex versus XX/XY complement) and shows that having two X chromosomes (rather than XY), on its own, can extend survival. (Note however that this does not replace human data as it does not explain how genetic variation impacts longevity.) Related to this, across 229 species the homogametic sex outlives the heterogametic sex by ~18% on average.
One interesting study examined the structure of protein-protein interactions, grouped by the genomic location of the underlying gene by chromosome. Here, they found that chromosomes 4, 13, 21, X, and Y are “less connected” than the rest of the chromosomes, suggesting one possible reason some whole-chromosome aneuploidies are more compatible with survival than others

Even subtle, acquired dosage changes track survival: men who mosaically lose the Y chromosome in blood (mLOY) show higher mortality and cardiovascular and cancer risk, see “Loss of Y”.
Genetics can also be used to not just understand causal effects of genetic variants or DNA structure, but also intermede dynamic phenotypes via Mendelian randomisation. MR uses inherited genetic variation as a kind of natural experiment to infer whether an exposure causally affects an outcome. Within drug discovery, it can tell us how a biomolecule of interest (e.g., a blood-based protein) drives disease pathology whilst “regressing out” genetic differences.
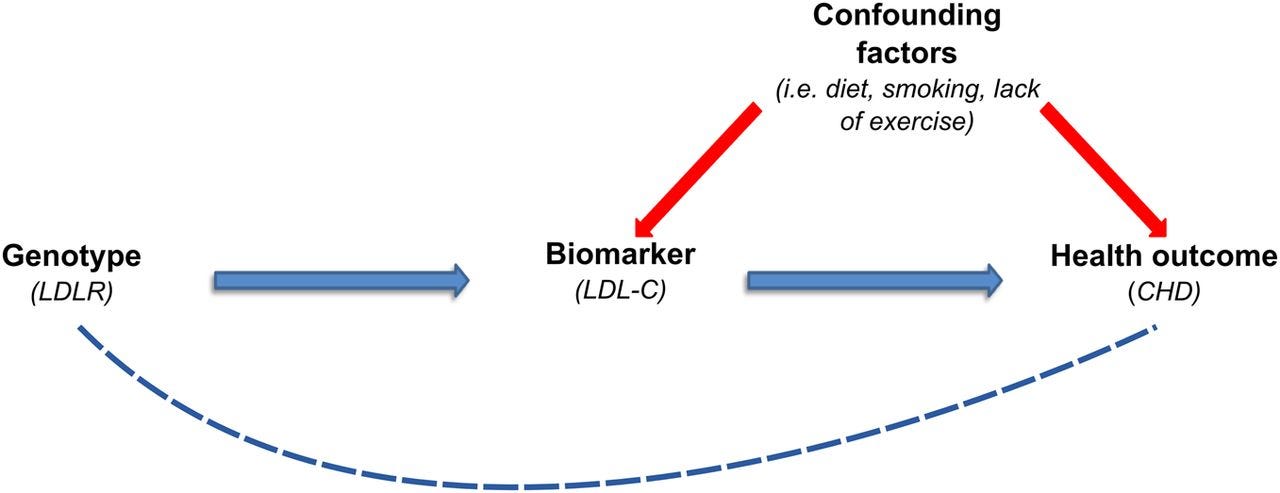
Aside, note that the X chromosome itself has historically been under-analysed in GWAS because of technical and statistical complications.